Like any complex machine comprised of multifarious but highly synchronized parts, cancer is the creation of diverse but organized cells and molecules driven by disinformation and dysfunction to produce malignant tissues and tumors.
Our program focuses on determining how nutrients and the microenvironment surrounding tumors interact to direct the formation, growth and spread of cancer cells. From individual metabolites to stromal cells, such as fibroblasts and immune cells, our goal is to discern the basic workings of tumors that cause disease, revealing a more comprehensive view of cancer biology that can be a guide to new therapies.
Director’s Statement
“We live and work in the tumor ecosystem, probing its nooks, crannies and complexities for new insights into how tumors begin and grow—and how best to prevent or stop both. We study paramount processes in cancer, including metabolism, signal transduction, tumor-stroma crosstalk and molecular machines. We want to know what different parts of a tumor do, how they work alone or together and why so that we can, ultimately, fix the problems that result in cancer. ”

 May 22, 2026
May 22, 2026 Mar 30, 2026
Mar 30, 2026 Mar 16, 2026
Mar 16, 2026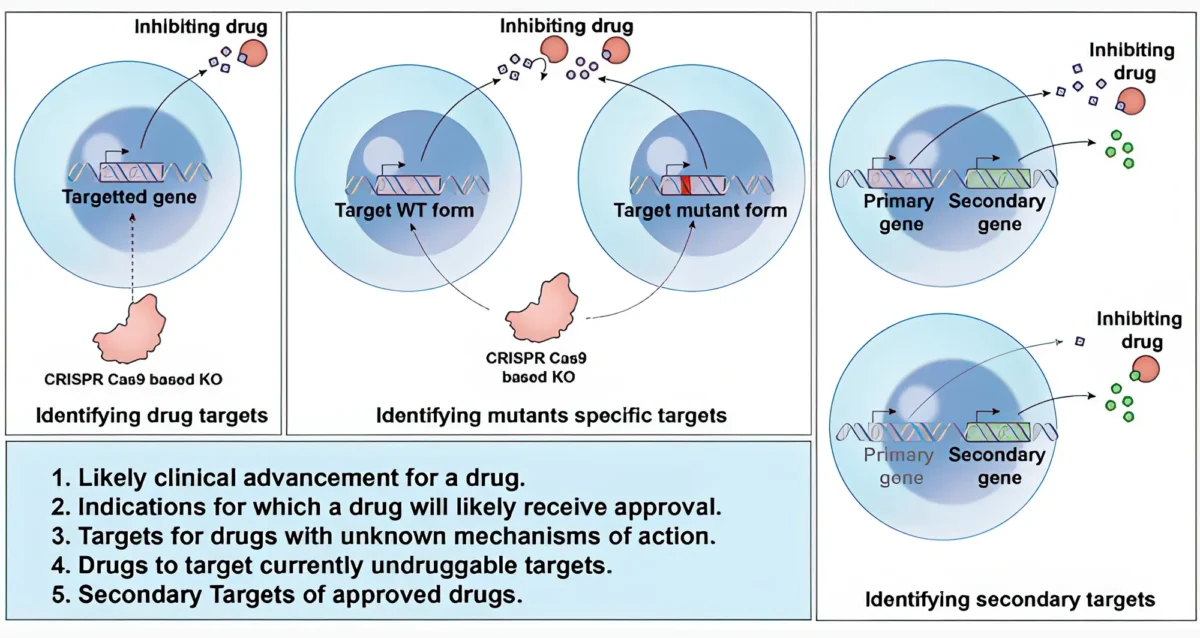 Nov 14, 2025
Nov 14, 2025 Nov 10, 2025
Nov 10, 2025 Oct 21, 2025
Oct 21, 2025