Dr. Freeze earned his PhD from the University of California, San Diego in 1976. Subsequently he held fellowships in Biology, Medicine and Neurosciences later joined the faculty at the same institution. In 1988 Dr. Freeze was recruited to Sanford Burnham Prebys.
Related Disease
Cancer, Congenital Disorders of Glycosylation, Crohn’s Disease (Colitis), Glycosylation-Related Disorders
Dr. Freeze’s research focuses on the pathology resulting from faulty glycosylation, the process of adding carbohydrate (sugar) chains to proteins and lipids. Carbohydrates are required for proper secretion and targeting of thousands of proteins – an often over looked fact of biology. He is driven by the search for novel therapeutics to treat patients with mutations leading to glycosylation defects called Congenital Disorders of Glycosylation (CDG).
Hudson Freeze’s Research Report
Glycosylation: An Essential Function
The entire cell surface is coated with sugars in complex chains that promote (or sometimes interfere) with cell-to-cell communication. These sugar chains are first attached to proteins deep inside the cell where they help them get into shape for their jobs. As the proteins percolate toward to cell surface and beyond, the sugar chains are sculpted for specific needs. This entire process, called glycosylation, recruits a force of more than 500 genes for this job. The Freeze lab works on several facets of glycosylation, all of them with an eye toward therapeutic applications for diseases that impair the functions of these critical genes.
Human Glycosylation Disorders
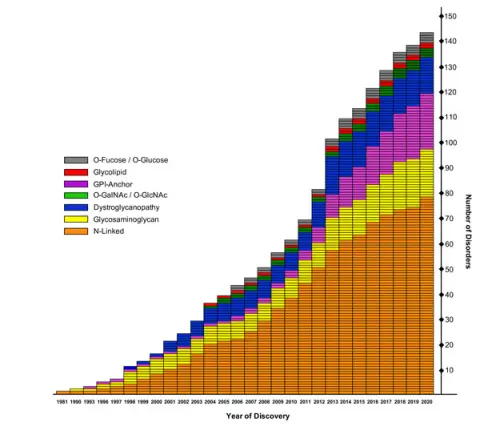
We focus is on a group of metabolic diseases called Congenital Disorders of Glycosylation (CDG). Today we know of defects in over 140 genes, well over double those known only 10 years ago. Patients with these diseases have highly variable mental and motor developmental delay, seizures, failure to grow, hypoglycemia (low blood sugar), clotting and digestion abnormalities and skeletal abnormalities to name just a few. These are rare disorders having a few thousand known patients worldwide, but it is likely that many remain undiagnosed, especially in developing countries.
Growing Awareness Physicians are becoming more aware of glycosylation disorders in general, and basic scientists continue to discover sugar chains at the helm of many basic metabolic processes. Defective glycosylation is also known to cause 15 types of muscular dystrophy.
The explosive growth in the number of different diseases caused by defective glycosylation.
Working with CDG Kids Rocket: We helped diagnose this young man over 10 years ago, and although he tragically passed away, his gentle face still reminds us of who we work for.
Rocket Williams reaches out to us. Learn more about the Rocket Fund.
Brianna: Sometimes our work leads to brighter outcomes as in the case of Brianna who we met over 20 years ago and treated with a simple therapy.
Flourishing, Brianna is now training to become a veterinarian.
Our Collaborations The Freeze lab not only identifies new glycosylation disorders, but also tries to understand how these defects cause the disease manifestations. Defects occur in genes that activate and transport sugars, assemble them into glycans and remodel them. Some also traffic and distribute the glycosylation machinery within cells. Ongoing collaborations with academic physicians provide a steady flow of new patients for analysis. Since very few laboratories in the United States work on CDG, we are developing new molecular diagnostic methods to handle the increasing number of patients. Increased awareness of CDG in the medical community generated expanded government funding that allies us with 10 medical centers in the US seeking additional patients to study their natural history, develop biomarkers and test emerging therapies. Those avenues along with the help of generous philanthropic support, enables us to extend efforts to supplement the depleted glycosylation pathways in patients.
In this image and film clip above, Harrison Ford poses a few questions for us.